Performance and Free Energy Estimation for
Solvated Polypeptides and Proteins Using Partial
Infinite Swapping
Download a PDF here and supplementary information here.
The method is more extensively detailed in my PhD thesis, Chapter 3
Partial infinite swapping (PINS) is a powerful enhanced sampling method for complex systems. In the present work thermodynamic observables are determined from reweighting at the post-processing stage for folding of (Ala)_10 in implicit and explicit solvent and for Xenon migration in myoglobin.


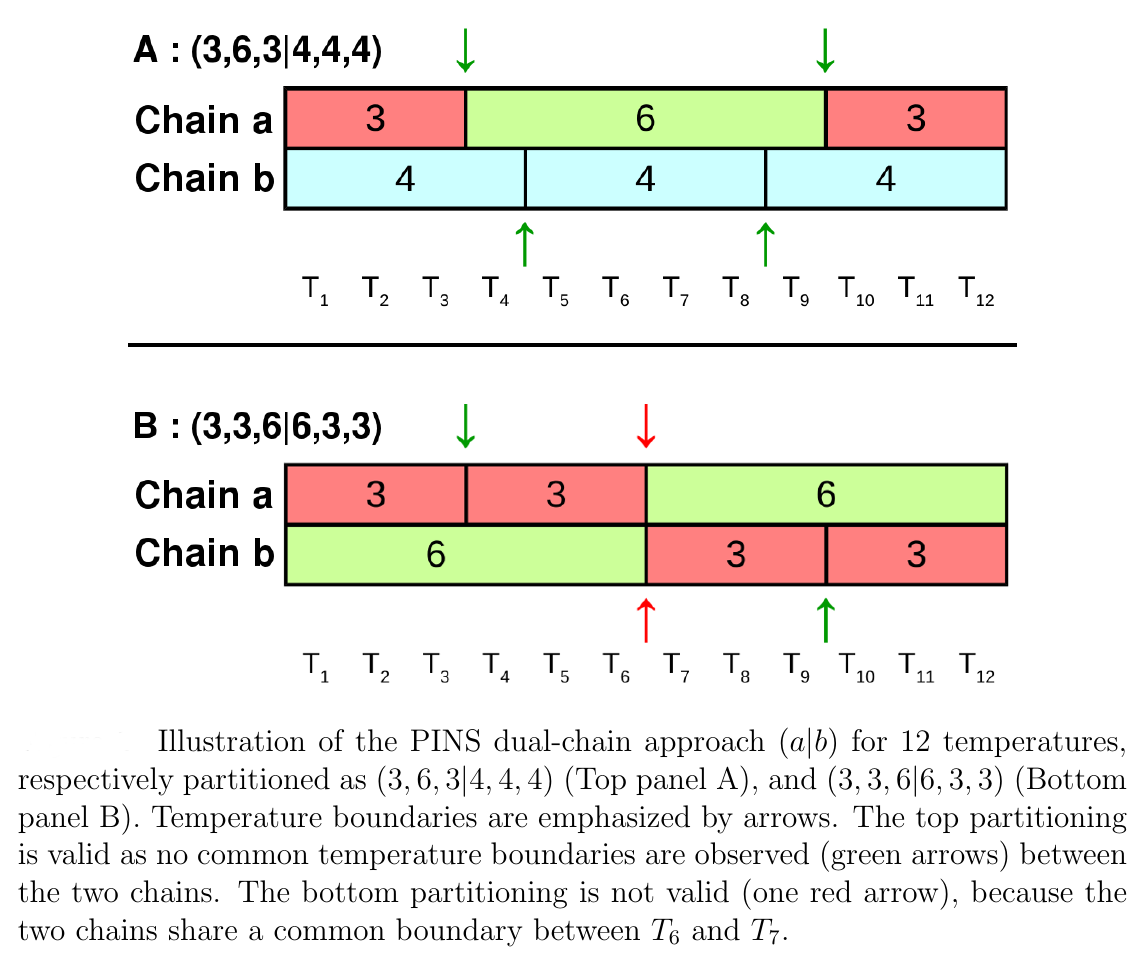
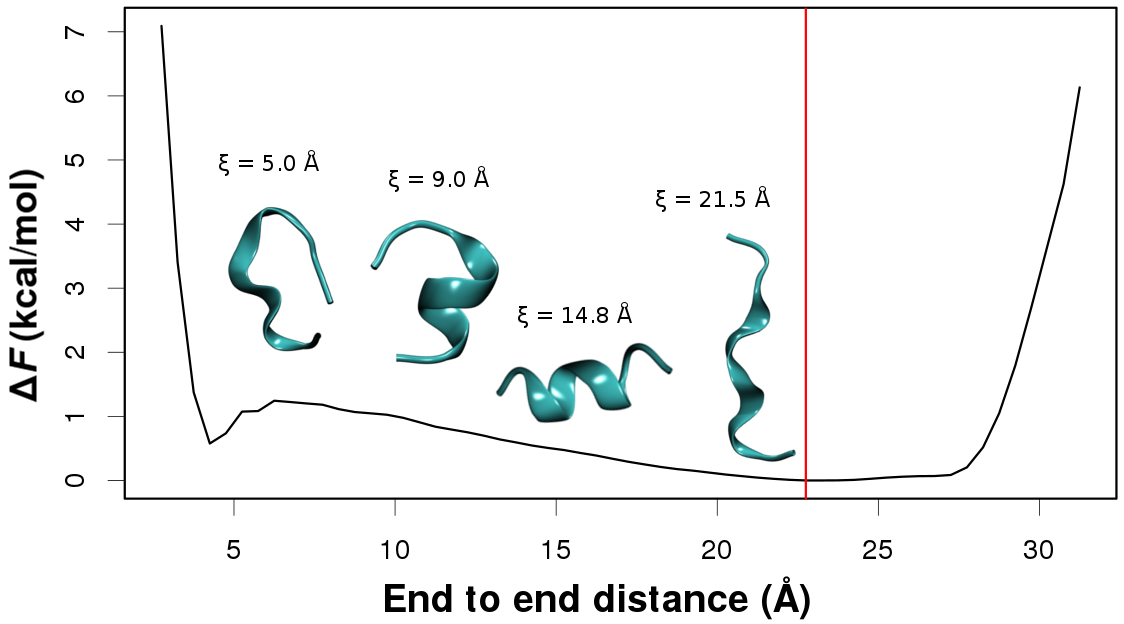
In every case free energy surfaces are determined using PINS with an accuracy comparable to Molecular Dynamics and Parallel Tempering simulations but at considerably reduced computational cost. Round trip times through the ensemble of temperature space are shown to be almost one order of magnitude shorter for PINS compared to PT simulations for (Ala)_10 in implicit solvent which suggests that PINS is more efficient for sampling diverse structures. Consistent with NMR experiments on shorter (Ala)_7 poly-alanine peptides, simulations of (Ala)_10 in explicit solvent highlight the essential role played by the environment in stabilizing extended conformations which are unfavourable in implicit solvent. Additional low-energy regions are beta-hairpins, 1 to 2 kcal/mol above the minimum energy structure.

For Xenon migration in Myoglobin, PINS finds stabilization energies of the experimentally known Xenon-pockets to range from -4.6 to -6.2 kcal/mol, in accord with experiment. Furthermore, the barrier heights between neighboring pockets have been determined to be ~ 4 kcal/mol.

By starting simulations from individual pockets, PINS finds sampling of the entire pocket network on the time scale of 3 ns using 32 replicas whereas with MD, 100 ns are not sufficient to access all pockets. Hence, PINS with reweighting is found to be both, a quantitatively accurate and computationally efficient method for studying complex biological molecules in solution.
See the page “Contributions to CHARMM” for details concerning the CHARMM implementation.