See arXiv: 1807.02431 for a preprint, or download it here, or browse a (slightly older) HTML version here
Final published version available in Computer Physics Communications , pdf available here
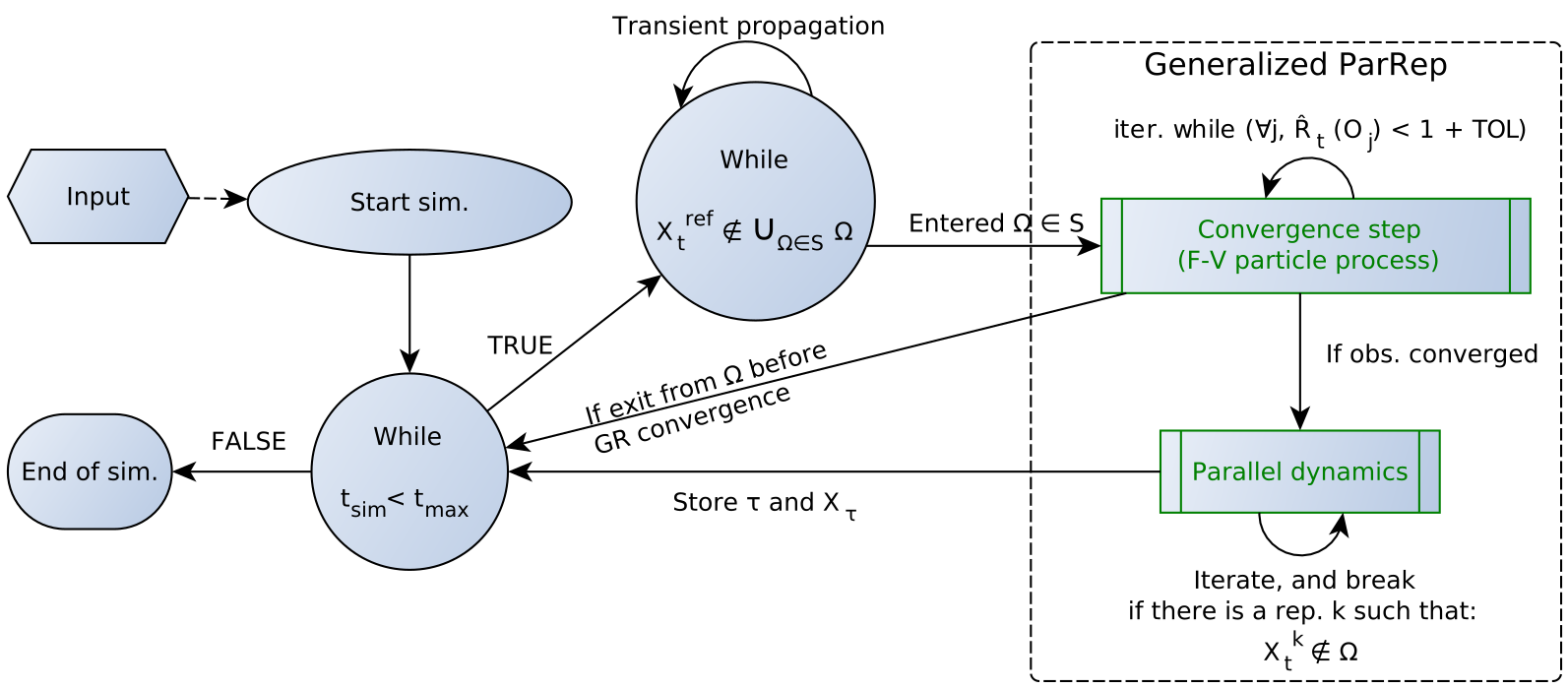
Metastability is one of the major encountered obstacle when performing long molecular dynamics simulations, and many methods were developed to address this challenge. The “Parallel Replica”(ParRep) dynamics is known for allowing to simulate very long trajectories of metastable Langevin dynamics in the materials science community, but it relies on assumptions that can hardly be transposed to the world of biochemical simulations.
The later developed “Generalized ParRep” variant solves those issues, but it was not applied to significant systems of interest so far.

In this article we present a new implementation of the Generalized Parallel Replica method, targeting frequently encountered metastable biochemical systems, such as conformational equilibria (validation with alanine dipeptide) or dissociation of protein-ligand complexes (study of the FKBP-DMSO system).
It will be shown that the resulting software exhibits a strong linear scalability, providing up to 70 % of the maximum possible speedup on several hundreds of CPUs.