See [1]
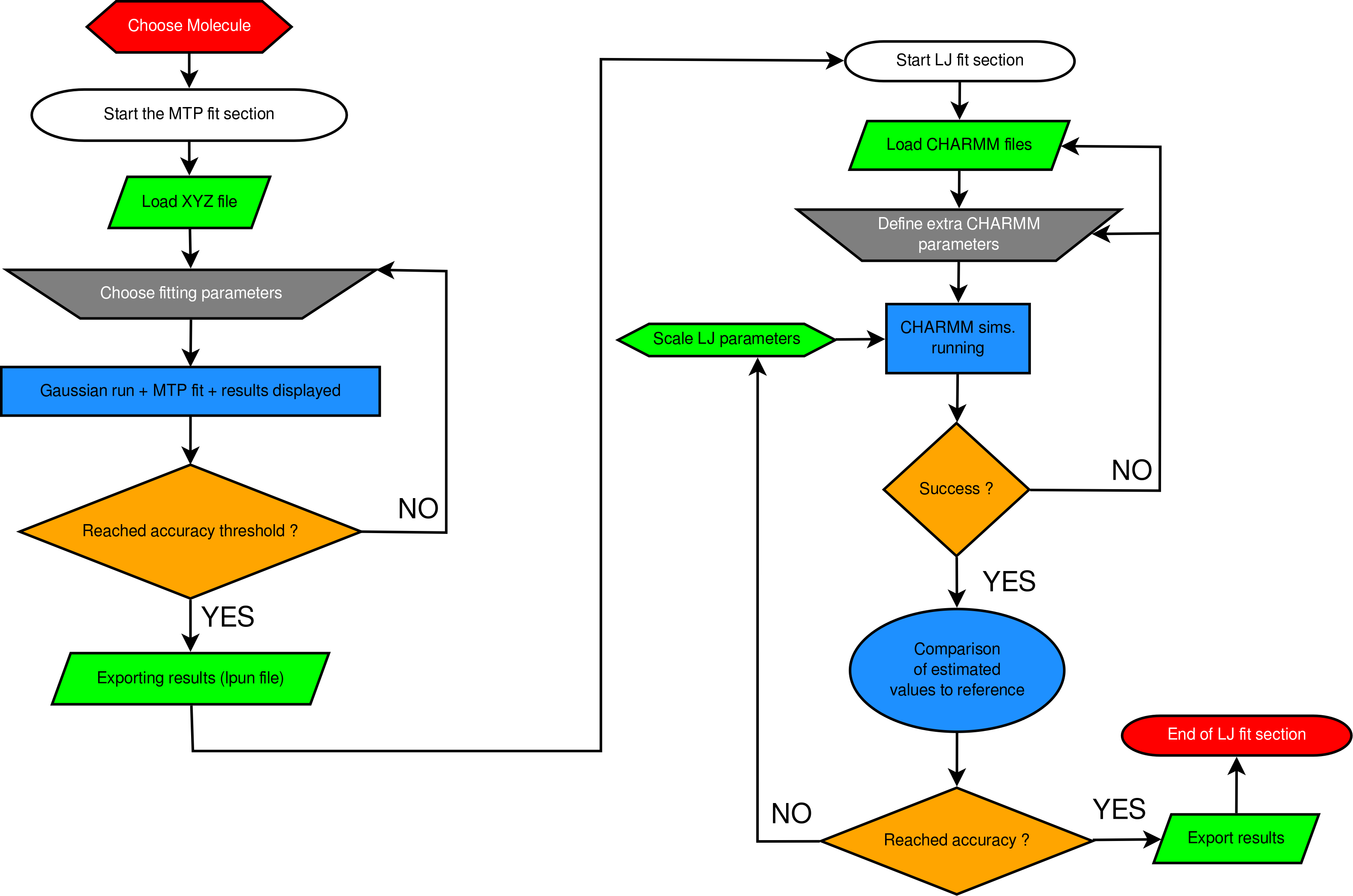
The quality of atomistic simulations depends decisively on the accuracy of the underlying energy function (force field). Of particular importance for condensed-phase properties are nonbonded interactions, including the electrostatic and Lennard-Jones terms. Permanent atomic multipoles (MTPs) are an extension to common point-charge (PC) representations in atomistic simulations. MTPs are commonly determined from and fitted to an ab initio Electrostatic Potential (ESP), and Lennard-Jones (LJ) parameters are obtained from comparison of experimental and computed observables using molecular dynamics (MD) simulations. For this a set of thermodynamic observables such as density, heat of vaporization, and hydration free energy is chosen, to which the parametrization is fitted.
The current work introduces a comprehensive computing environment (Fitting Wizard (FW)) [1] for optimizing nonbonded interactions for atomistic force fields of different qualities. The FW supports fitting of standard PC-based force fields and more physically motivated multipolar (MTP) force fields.
[1] Florent Hédin, Krystel El Hage, and Markus Meuwly, J. Chem. Inf. Model., 2016, 56 (8), pp 1479–1489
For instructions concerning download, installation and use, see the following:
https://github.com/MMunibas/FittingWizard
https://github.com/MMunibas/FittingWizardWeb